Statistics
Misc
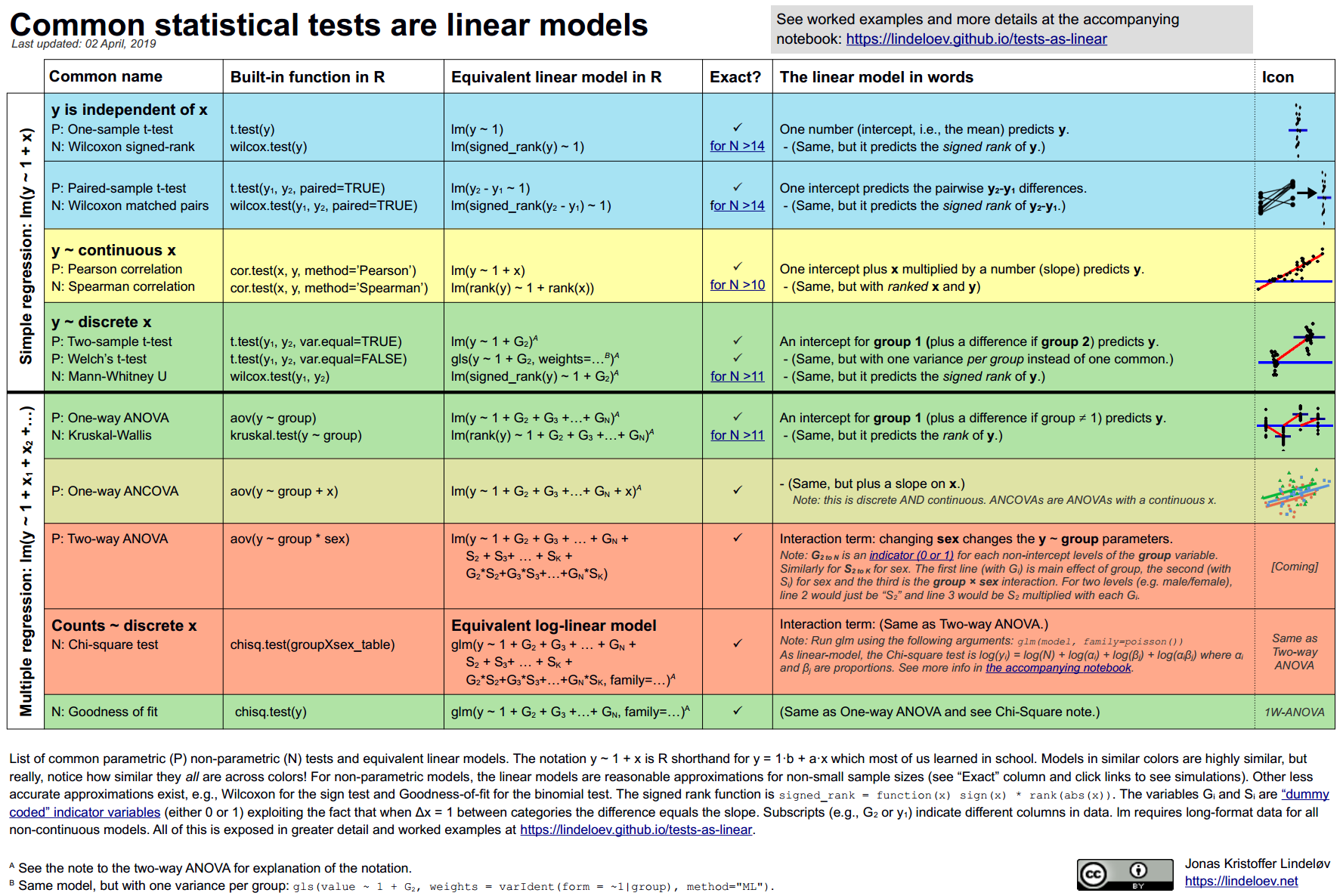
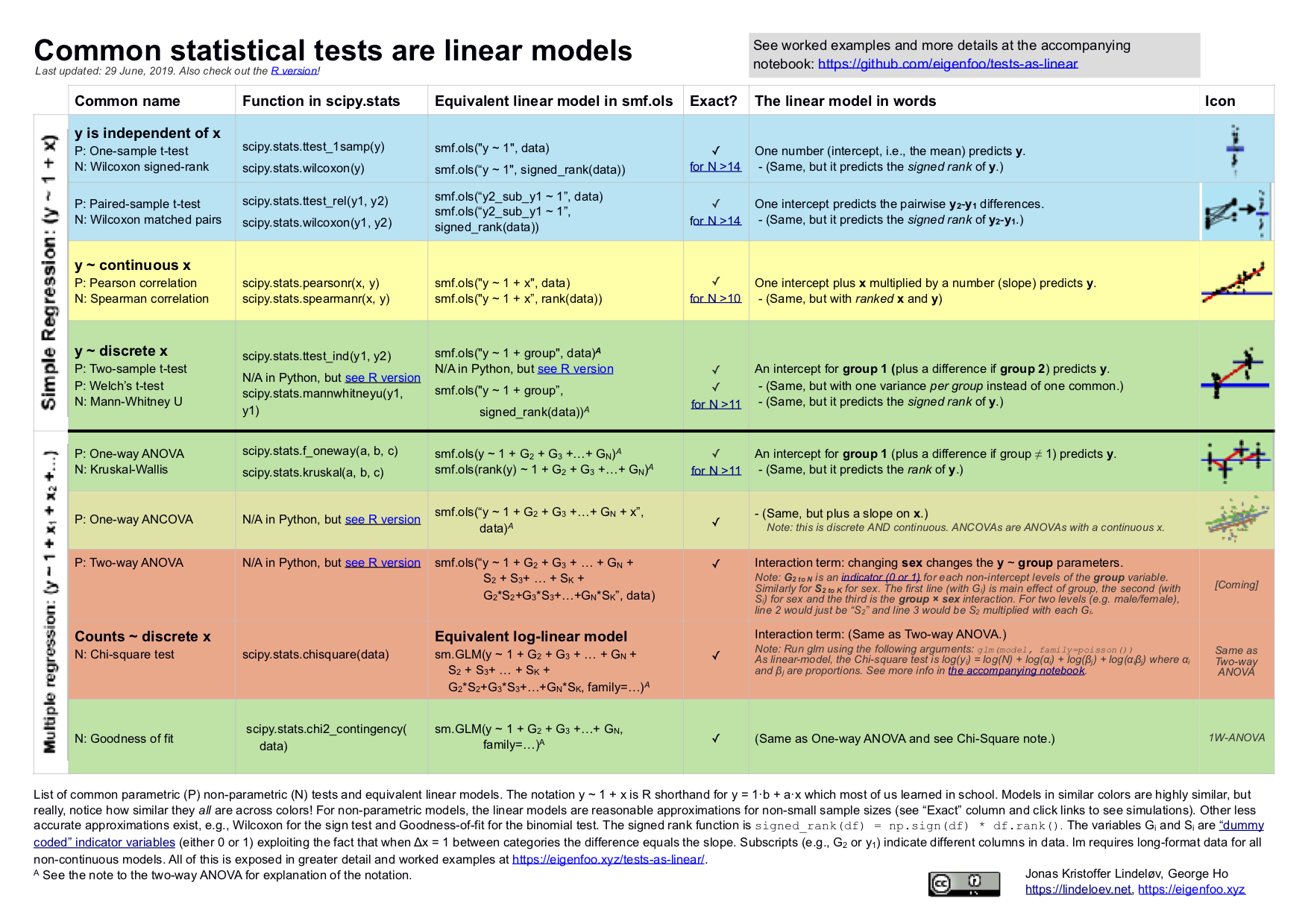
- Common Statistical Tests are Linear Models
- Papers
- Age Adjustment of per 100K rate
.png)
- Allows communities with different age structures to be compared
- The crude (unadjusted)1994 cancer mortality rate in New York State is 229.8 deaths per 100,000 men. The age-adjusted rate is 214.7 deaths per 100,000 men.
- Notice that 214.7 isn’t 229.8*1 of course but the sum of all the individual age-group weighted rates.
- Process (Formulas in column headers)
- Calculate the disease’s rate per 100K for each age group
- Multiply the age-specific rates of disease by age-specific weights
- The weights are the proportion of the US population within each age group. (e.g. 0-14 year olds are 28.4% of the 1970 US population)
- The weighted rates are then summed over the age groups to give the (total) age-adjusted rate
Terms
Bias-Variance Tradeoff - Describes the balance between two types of errors that affect a model’s performance. Increasing one, decreases the other which affects your model’s ability to generalize to new data. (i.e. underfitting vs overfitting)
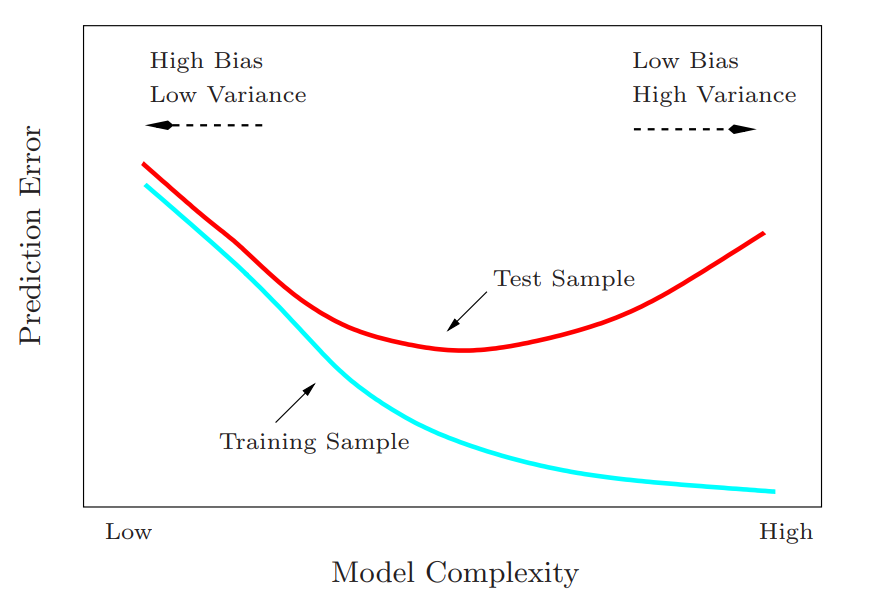
Characterized by Mean Squared Error
\[\begin{align} \text{MSE} &= \mbox{Variance} + \mbox{Bias}^2 + \text{noise}\\ &= \mbox{Var}(y - \hat y) + (\mbox{mean}(y) - \mbox{mean}(\hat y))^2 + \mbox{Var}(\epsilon) \end{align}\]
Bias: The error introduced by approximating a real-world problem, which may be complex, by a simplified model. High bias can cause a model to miss relevant patterns, leading to systematic errors and underfitting. Also see Mathematics, Glossary >> Bias
Variance: The error introduced by the model’s sensitivity to small fluctuations in the training data. High variance can cause a model to model the random noise in the training data, leading to overfitting and poor generalization to new data.
In extreme cases,
- \(\uparrow\) Variance, \(\downarrow\) Bias - Think “reducing \(\epsilon\) in linear regression”; The model explains more of the variance in the (training) data, but it’s more prone to overfitting.
- \(\downarrow\) Variance, \(\uparrow\) Bias - The model doesn’t explain much of the variance in the (training) data, and it doesn’t generalize well (i.e. underfits).
Normally distributed residuals (aka random noise) indicates a high variance low bias model since no information is left in the data that is unexplained by the model.
Coefficient of Variation (CV) - aka Relative Standard Deviation (RSD) - aka Dispersion Parameter - Measure of the relative dispersion of data points in a data series around the mean. Usually expressed as a percentage.
\[CV = \frac{\sigma}{\mu}\]
While most often used to analyze dispersion around the mean, a quartile, quintile, or decile CV can also be used to understand variation around the median or 10th percentile, for example.
Should only be used with variables that have minimum at zero (e.g. counts, prices) and not interval data (e.g celsius or fahrenheit)
When the mean value is close to zero, the coefficient of variation will approach infinity and is therefore sensitive to small changes in the mean. This is often the case if the values do not originate from a ratio scale.
A more robust possibility is the quartile coefficient of dispersion, half the interquartile range divided by the average of the quartiles (the midhinge)
\[CV_q = \frac{0.5(Q_3 - Q_1)}{0.5(Q_3 + Q_1)}\]
For small samples (Normal Distribution)
\[CV_*= (1 + \frac{1}{4n}) CV\]
For log-normal distribution
\[CV_{LN} = \sqrt{e^{\sigma^2_{ln}} - 1}\]
- Where \(\sigma_{ln}\) is the standard deviation after a \(\ln\) transformation of the data
Covariance - Between two random variables is a measure of how correlated are their variations around their respective means
Empirical Size - Rejection rate of a test under the null in a simulation or experimental conditions
- In the context of hypothesis testing via Monte Carlo simulations refers to the proportion of times that the null hypothesis is incorrectly rejected across the simulations when the null hypothesis is true. This is essentially a simulated version of a Type I error rate.
- Example: (source) For a nominal rejection rate of \(\alpha\) = 0.05, it a test under a null distribution has:
- Empirical Size > 0.05, it’s too liberal, i.e. rejects the null too much when it’s true
- Empirical Size < 0.05, its too conservative, i.e. doesn’t reject the null enough when it’s true
- The difference to 0.05 is commonly called size distortion
Estimand, Estimator, Estimate (source)
- Estimand - Parameter in the population which is to be estimated in a statistical analysis
- Estimator - A rule for calculating an estimate of a given quantity based on observed data
- Function of the observations, i.e., how observations are put together
- e.g. linear regression function, variance estimator (\(s^2 = \sum (x_i - \bar x)^2/(n-1)\))
- Estimate - An approximation, which is a value that is usable for some purpose even if input data may be incomplete, uncertain, or unstable (value derived from the best information available)
Grand Mean - The mean of group means
- Use Cases
- Hierarchical Group Comparison: Allows you to see how the average of each group relates to the overall average across all groups.
- Example: Imagine you collect data on the growth of various species of plants in different types of soil. Calculating the grand mean for plant height of each species allows you to see if any specific soil type consistently produces taller plants compared to the overall average.
- Identifying Outliers: If the mean of a specific group deviates significantly from the grand mean, it might indicate the presence of outliers in that group.
- ANOVA: It provides a baseline against which the individual group means are compared to assess if there are statistically significant differences between them.
- Hierarchical Group Comparison: Allows you to see how the average of each group relates to the overall average across all groups.
- It’s the intercept value when you use Sum-to-Zero (used in ANOVA) or Deviation contrasts (See Regression, Linear >> Contrasts >> Sum-to-Zero, Deviation)
- Use Cases
Kernel Smoothing - Essence is the simple concept of a local average around a point, x; that is, a weighted average of some observable quantities, those of which closest to x being given the highest weights
Margin of Error (MoE) - The range of values below and above the sample statistic in a confidence interval.
\[\text{MOE}_\gamma = z_\gamma \sqrt{\frac{\sigma^2}{n}}\]
- MOE is the z-score with confidence level γ ⨯ Standard Error
- In general, for small sample sizes (under 30) or when you don’t know the population standard deviation, use a t-score to get the critical value. Otherwise, use a z-score.
- See Null Hypothesis Significance Testing (NHST) >> Misc >> Z-Statistic Table for an example
- Example: a 95% confidence interval with a 4 percent margin of error means that your statistic will be within 4 percentage points of the real population value 95% of the time.
- Example: a Gallup poll in 2012 (incorrectly) stated that Romney would win the 2012 election with Romney at 49% and Obama at 48%. The stated confidence level was 95% with a margin of error of ± 2. We can conclude that the results were calculated to be accurate to within 2 percentages points 95% of the time.
- The real results from the election were: Obama 51%, Romney 47%. So the Obama result was outside the range of the Gallup poll’s margin of error (2 percent).
- 48 – 2 = 46 percent
- 48 + 2 = 50 percent
- The real results from the election were: Obama 51%, Romney 47%. So the Obama result was outside the range of the Gallup poll’s margin of error (2 percent).
Median Absolute Deviation (MAD) - The median of the absolute deviations from the data’s median. A robust measure of the variability of a univariate sample of quantitative data. Typically used as a substitute for the standard deviation in standardization.
\[\text{MAD} = \text{median}(\;|X_i - \text{median}(\boldsymbol{X})|\;)\]
Normalization - Rescales the values into a specified range, typically [0,1]. This might be useful in some cases where all parameters need to have the same positive scale. However, the outliers from the data set are lost.
\[\tilde{X} = \frac{X-X_{\text{min}}}{X_{\text{max}}-X_{\text{min}}}\]
- Some functions have the option to normalize with range [-1, 1]
- Best option if the distribution of your data is unknown.
Parameter - Describes an entire population (also see statistic)
P-Value - \(\text{p-value}(y) = \text{Pr}(T(y_{\text{future}}) >= T(y) | H)\)
- \(H\) is a “hypothesis,” a generative probability model
- \(y\) is the observed data
- \(y_{\text{future}}\) are future data under the model
- \(T\) is a “test statistic,” some pre-specified specified function of data
Sampling Distribution - For the mean of a sample, it will vary from sample to sample; the way this variation occurs is described by the “sampling distribution” of the mean.
- Applies to other quantities that may be estimated from a sample, such as a proportion or regression coefficient, and to contrasts between two samples, such as a risk ratio or the difference between two means or proportions. All such quantities have uncertainty due to sampling variation, and for all such estimates a standard error can be calculated to indicate the degree of uncertainty.
Sampling Error - The difference between population parameter and the statistic that is calculated from the sample (such as the difference between the population mean and sample mean).
Standard Deviation - The standard deviation of a sample is an estimate of the variability of the population from which the sample was drawn.
Standard Error - For means, it’s a calculation of how much sample means will vary using the standard deviation of its sampling distribution, which we call the standard error (SE) of the estimate of the mean, i.e. a measure of the precision or uncertainty of the sample mean.
Standard Error of the Mean (SEM)] (Also see Descriptive Statistics >> Interpreting s.d., s.e.m, and CI Bars)
\[\text{SEM} = \frac{\text{SD}}{\sqrt{n}}\]
- Assumes a simple random sample with replacement from an infinite population
Standardization rescales data to fit the Standard Normal Distribution which has a mean (μ) of 0 and standard deviation (σ) of 1 (unit variance).
\[\tilde X = \frac{X-\mu}{\sigma}\]
- Recommended for PCA and if your data is known to come from a Gaussian distribution.
Statistic - Describes a sample (also see parameter)
Variance (\(\sigma^2\))- Measures variation of a random variable around its mean.
Variance-Covariance Matrix - Square matrix containing variances of the fitted model’s coefficient estimates and the pair-wise covariances between coefficient estimates.
\[\begin{align} &\text{Cov}(\hat\beta) = (X^TX)^{-1} \cdot \text{MSE}\\ &\text{where}\;\; \text{MSE} = \frac{\text{SSE}}{\text{DSE}} = \frac{\text{SSE}}{n-p} \end{align}\]
Diagnonal is the variances, and the rest of the values are covariances
There’s also a variance/covariance matrix for error terms
Example
x <- sin(1:100) y <- 1 + x + rnorm(100) MSE <- sum(residuals(lm(y ~ x))^2)/98 # where 98 is n-2 vcov_mat <- MSE * solve(crossprod(cbind(1, x)))- vcov_mat is the same as
vcov(lm(y ~ x))
- vcov_mat is the same as
Null Hypothesis Significance Testing (NHST)
Misc
- Papers
- Rethinking Type S and Type M Errors (Lakens et al 2026)
- “Instead of reporting Type S error rates, researchers should perform tests against a range of values considered theoretically or practically equivalent to 0. Instead of reporting Type M errors, researchers should report bias-corrected effect-size estimates provided by methods such as p-uniform and report the critical effect size.”
- Rethinking Type S and Type M Errors (Lakens et al 2026)
- Why would we not always use a non-parametric test so we do not have to bother about testing for normality? The reason is that non-parametric tests are usually less powerful than corresponding parametric tests when the normality assumption holds. Therefore, all else being equal, with a non-parametric test you are less likely to reject the null hypothesis when it is false if the data follows a normal distribution. It is thus preferred to use the parametric version of a statistical test when the assumptions are met.
- If your statistic value is greater than the critical value, then it’s significant and you reject H0
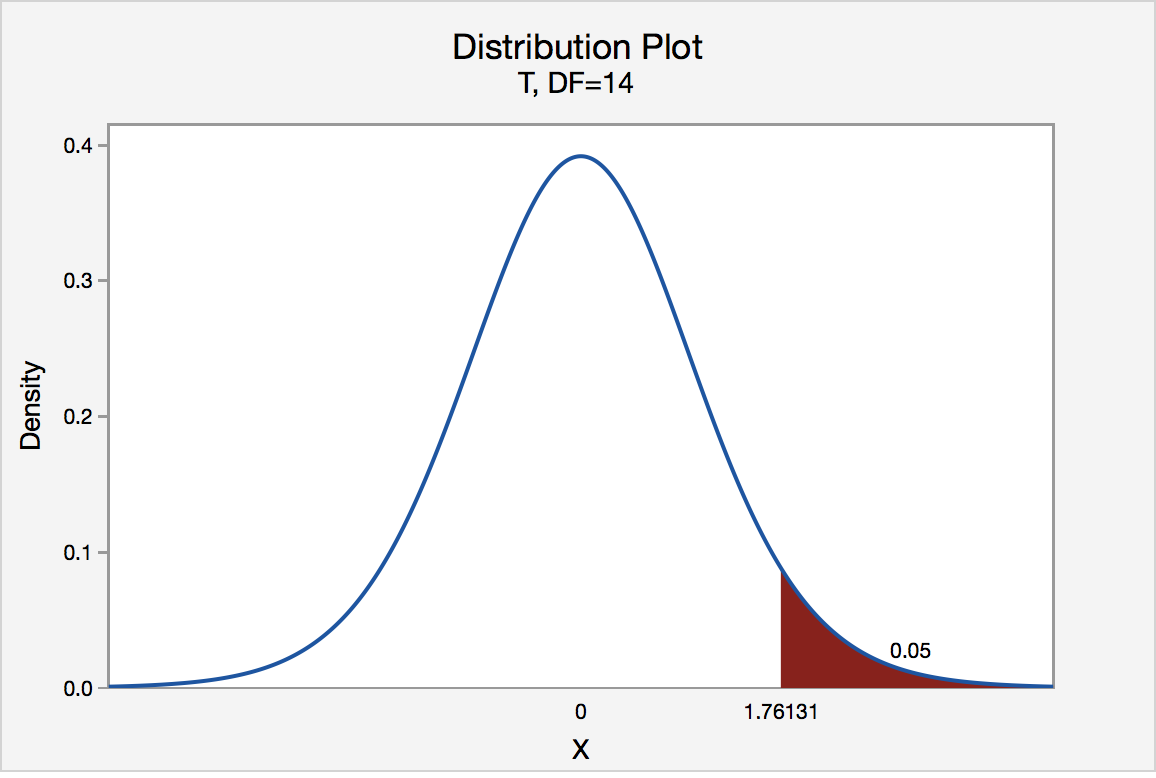
- Think in terms of a distribution with statistic values on the x-axis, and greater than the critical value means you’re in the tail (one-sided)
- T-Statistic Table
- Z-Statistic Table
- Example: 95% CI → α = 100% - 95% = 0.05 → α/2 (1-tail) = 0.025
- 1 - 0.025 = 0.975 (subtract from 1 because the z-score table cells are for the area left of the critical value
- The z-score is 1.96 for a 95% CI
.png)
- Z-score comes from adding the row value with the column value that has the cell value of our area (e.g. 0.975) left of the critical value
- If the area was between 0.97441 and 0.97500, then the z-score would be the row value, 1.9, added to the column value that’s half way between 0.05 and 0.06, which results in a z-score of 1.955
- Example: 95% CI → α = 100% - 95% = 0.05 → α/2 (1-tail) = 0.025
Terms
- Exact Tests - Statistical tests where the p-value is computed directly from the exact probability distribution of the test statistic under the null hypothesis, rather than from a large-sample approximation (aka asymptotic tests). Asymptotic tests approximate using distributions such as the normal, chi-squared, or t-distribution.
- Exact tests construct the distribution of the test statistic by enumerating all possible outcomes that could have been observed under the null hypothesis, given the fixed features of the data (e.g., sample size, marginal totals).
- Guarantees Type I Error Control: Exact tests ensure the significance level (α) is never exceeded — the probability of a false positive is at most α, not approximately α. Asymptotic tests can exceed α in small samples, which is a serious flaw in confirmatory settings.
- Handles Small Sample Sizes: Exact tests compute probabilities directly from the data’s actual distribution, giving valid results regardless of sample size.
- Handles Sparse or Unbalanced Data: When cell counts in contingency tables are low (e.g., expected frequencies below 5), asymptotic tests like the chi-squared test lose accuracy.
- Computationally intensive for larger data sets.
- Power - 1-β where beta is the Probability of a type II error
- Simultaneity - A set of hypothesis tests is simultaneous when you want your inferential guarantee to hold jointly across all tests at once, rather than individually for each one.
Errors and Rates
Type I Error - False-Positive; occurs if an investigator rejects a null hypothesis that is actually true in the population
- The models perform equally well, but the A/B test still produces a statistically significant result. As a consequence, you may roll out a new model that doesn’t really perform better.
- You can control the prevalence of this type of error with the p-value threshold. If your p-value threshold is 0.05, then you can expect a Type I error in about 1 in 20 experiments, but if it’s 0.01, then you only expect a Type I error in only about 1 in 100 experiments. The lower your p-value threshold, the fewer Type I errors you can expect.
Type II Error - False-Negative; occurs if the investigator fails to reject a null hypothesis that is actually false in the population
- The new model is in fact better, but the A/B test result is not statistically significant.
- Your test is underpowered, and you should either collect more data, choose a more sensitive metric, or test on a population that’s more sensitive to the change.
Type S Error (Sign Error): The A/B test shows that the new model is significantly better than the existing model, but in fact the new model is worse, and the test result is just a statistical fluke. This is the worst kind of error, as you may roll out a worse model into production which may hurt the business metrics.
- {retrodesign} - Provides tools for working with Type S (Sign) and Type M (Magnitude) errors. (Vignette)
Type M error (Magnitude Error): The A/B test shows a much bigger performance boost from the new model than it can really provide, so you’ll over-estimate the impact that your new model will have on your business metrics.
- {retrodesign} - Provides tools for working with Type S (Sign) and Type M (Magnitude) errors. (Vignette)
False Positive Rate (FPR)(\(\alpha\)) - ; Probability of a type I error; Pr(measured effect is significant | true effect is “null”)
\[\text{FPR} = \frac{v}{m_0}\]
- \(v\): Number of times there’s a false positive
- \(m_0\): Number of non-significant variables
False Discovery Rate (FDR) - Pr(measured effect is null | true effect is significant)
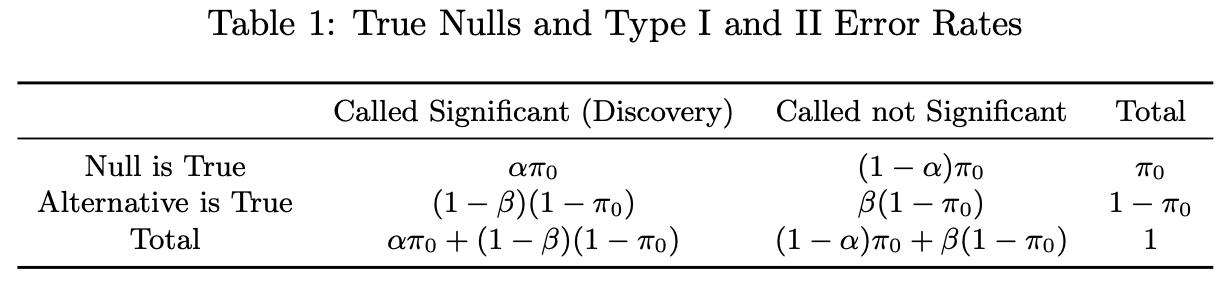
\[\text{FDR} = \frac{\alpha \pi_0}{\alpha \pi_0 + (1-\beta)(1-\pi_0)}\]
- \(\alpha\) - Type I error rate (False Positive Rate)
- \(\beta\) - Type II error rate (False Negative Rate)
- \(1-\beta\) - Power
- \(\pi_0\) - Count of true null effects
- \(1−\pi_0\) - Count of true non-null effects
Family-Wise Error Rate (FWER) - the risk of at least one false positive in a family of S hypotheses.
\[FWER = \frac{v}{R}\]
- \(v\): Number of times there’s a false positive
- \(R\): Number of times we claim β ≠ 0
- e.g. Using the same data and variables to fit multiple models with different outcome variables (i.e. different hypotheses)
- Tests belong in the same family when you want a collective guarantee — typically when:
They address a single research question together
A false positive in any one of them would be a problem for your conclusions
Multiple Testing Corrections
p.adjusthas most if not all the p-value multiple testing adjustment methodsBonferroni Correction
- Procedure
- Adjust p-values: \(p_i^{\text{adj}} = \min(1, m\cdot p_i)\)
- \(m\) is the number of experiments
- Reject Null if: \(p_i^{\text{adj}} \lt \alpha\)
- Adjust p-values: \(p_i^{\text{adj}} = \min(1, m\cdot p_i)\)
- Controls family-wise error rate (FWER).
- Guarantees the probability of a single false discovery rate ≤ α
- Higher rate of false negatives ⇒ Lower statistical power
- Zero risk tolerance
- With respect to Family-wise error rate (FWER) control, the Bonferroni correction can be conservative if there are a large number of tests and/or the test statistics are positively correlated.
- Procedure
Benjamini-Hochberg Correction
- Packages
- {eClosure} - Implements several methods for False Discovery Rate control based on the e-Closure Principle, in particular the Closed e-Benjamini-Hochberg and Closed Benjamini-Yekutieli procedures
- Procedure
- Sort your p-values: p₁ ≤ … ≤ pₘ.
- Accept the first k where pₖ > α/(m−k+1)
- Controls False Discovery Rate (FDR)
- Guarantees that among all discoveries, false positives are ≤ q
- Fewer false negatives ⇒ Higher statistical power
- Some risk tolerance
- Valid when the hypothesis tests are independent or when they are positively associated ( Sarkar, 1998; Sarkar and Chang, 1997).
- Hochberg p-values are faster to compute than Hommel’s.
- Packages
Benjamini-Yekutieli Correction
- Works like BH (step-up procedure on ordered p-values) but multiplies the BH critical values by a correction factor
- Controls FDR but handles correlated test statistics (joint distribution of test statistics)
Holm Correction
- Uniformly more powerful than Bonferroni and still controls FWER
- Handles correlated test statistics (joint distribution of test statistics)
Hommel Correction
- More powerful than Hochberg’s, but the difference is usually small and the Hochberg p-values are faster to compute.
- Valid when the hypothesis tests are independent or when they are positively associated ( Sarkar, 1998; Sarkar and Chang, 1997).
Romano and Wolf’s Correction
- Accounting for the dependence structure of the p-values (or of the individual test statistics) produces more powerful procedures than Bonferroi and Holms. This can be achieved by applying resampling methods, such as bootstrapping and permutations methods.
- Permutation tests of regression coefficients can result in rates of Type I error which exceed the nominal size, and so these methods are likely not ideal for such applications
- See Stata docs of the procedure
- Packages
- {wildrwolf}: Implements Romano-Wolf multiple-hypothesis-adjusted p-values for objects of type fixest and fixest_multi from the fixest package via a wild cluster bootstrap.
- Accounting for the dependence structure of the p-values (or of the individual test statistics) produces more powerful procedures than Bonferroi and Holms. This can be achieved by applying resampling methods, such as bootstrapping and permutations methods.
Permutation Testing
The main difference from typical NHST is that we create the distribution of the data under the null hypothesis using simulation instead of relying on a reference distribution (e.g. Normal).
Packages
- {coin} - Conditional inference procedures for the general independence problem including two-sample, K-sample (non-parametric ANOVA), correlation, censored, ordered and multivariate problems
- {modernBoot::perm_maxT} - The maxT method conducts individual t-tests for each variable, then corrects for multiple comparisons using the distribution of the maximum absolute t-statistic under permutation. This maintains FWER at the specified level while preserving power.
- {modernBoot::perm_text_2sample} - Performs exact or approximate permutation test for comparing two independent samples. Tests null hypothesis that two groups have equal distributions.
Assumes Exchangeability (See Mathematices, Glossary >> exchangeability)
Provided the sample sizes are approximately equal, the permutation test is robust against unequal variances (source)
General Procedure
- Calculate effect size (e.g. difference-in-means) of original data
- Permute group labels (i.e. treatment/control) (i.e. sample without replacement)
- Calculate the effect size of the permuted data and store it
- Repeat steps 2 and 3 about 10000 times to get the Null Distribution
- Calculate the percentage of times the effect size from the permuted data was more extreme than the effect size from the original data. This is the p-value.
- Reject the Null Hypothesis (no effect) if the p-value is less than \(\alpha\) (e.g. 0.05).
Example 1: Difference-in-Means (source)
# Generating data set.seed(2021) d <- as.data.frame(cbind(rnorm(1:20, 500, 50), c(rep(0, 10), rep(1, 10)))) treatment <- d$V2 outcome <- d$V1 #Difference in means original <- diff(tapply(outcome, treatment, mean)) mean(outcome[treatment==1]) - mean(outcome[treatment==0]) #Permutation test permutation.test <- function(treatment, outcome, n){ distribution <- c() result=0 for (i in 1:n) { distribution[i] <- diff(by(outcome, sample(treatment, length(treatment), FALSE), mean)) } result <- sum(abs(distribution) >= abs(original))/(n) # p-value return(list(result, distribution)) } (test1 <- permutation.test(treatment, outcome, 10000)) [1] 0.7163Visualization
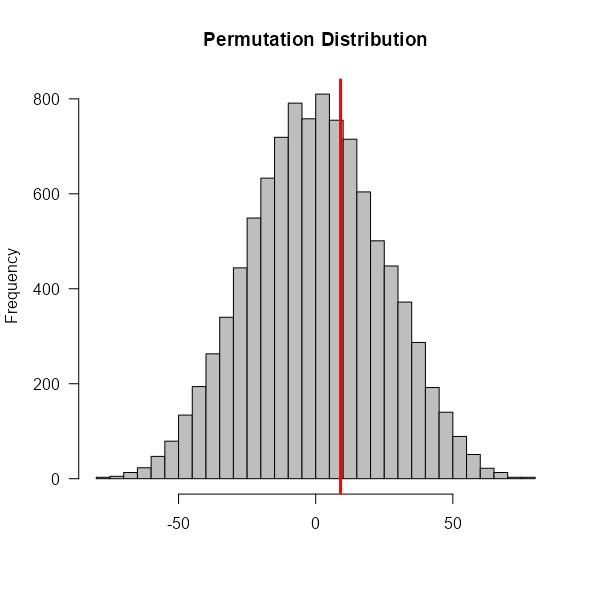
Code
hist(test1[[2]], breaks=50, col='grey', main="Permutation Distribution", las=1, xlab='') abline(v=original, lwd=3, col="red")
Example 2: Distance Correlation (source)
permutestdcor <- function(X, Y, nperm=10000){ # Carries out the permutation test on dCor. n <- NROW(X) # sample size results <- rep(NA,nperm) set.seed(123) for (i in seq_len(nperm)){ if(NCOL(Y) == 1){ results[i] <- n * energy::dcor(X,Y[sample(1:n)])^2 } else { results[i] <- n * energy::dcor(X,Y[sample(1:n),])^2 } } p <- n * energy::dcor(X,Y)^2 CDF <- ecdf(results) return(1 - CDF(p)) # p-value }- Uses a permutation test to get the significance of the distance correlation
- Utilizes the ecdf to get the p-value
- The score is squared and multiplied by the sample size. I’m not sure if this because of the distance correlation or if it’s required in general when using the ecdf method of getting the p-value.
Example 3: {coin} (source)
# same data as example 1 library(coin) independence_test(outcome ~ treatment) #> Asymptotic General Independence Test #> #> data: outcome by treatment #> Z = 0.37231, p-value = 0.7097 #> alternative hypothesis: two.sided
Monte Carlo Testing
Example: {spatstat} book, p. 190 (See Geospatial, Point Patterns >> Misc>> Resources)
library(spatstat) denRed <- density(redwood, bw.ppl, ns = 16) # ns arg for bw.ppl obsmax <- max(denRed) # observed max density (i.e. intensity) simmax <- numeric(99) lamRed <- intensity(redwood) # avg density winRed <- as.owin(redwood) # study area for(i in 1:99) { # samples a random spatial process for the given params Xsim <- rpoispp(lamRed, win = winRed) denXsim <- density(Xsim, bw.ppl, ns = 16) simmax[i] <- max(denXsim) } (pval <- (1 + sum(simmax > obsmax)) / 100) #> [1] 0.01- Determines whether a cluster of locations (i.e. area of high density/intensity) is significantly different than what can be expected from a random spatial process
Bootstrapping
- Misc
- Also see
- Resources
- When the bootstrap doesn’t work (Lumley)
- Do NOT bootstrap the standard deviation (article)
- Bootstrap is “based on a weak convergence of moments”
- if you use an estimate based standard deviation of the bootstrap, you are being overly conservative (i.e. you’re overestimating the sd and CIs are too wide)
- Bootstrapping uses the original, initial sample as the population from which to resample, whereas Monte Carlo simulation is based on setting up a data generation process (with known values of the parameters of a known distribution). Where Monte Carlo is used to test drive estimators, bootstrap methods can be used to estimate the variability of a statistic and the shape of its sampling distribution
- Use bias-corrected bootstrapped CIs (article)
- “percentile and BCa methods were the only ones considered here that were guaranteed to return a confidence interval that respected the statistic’s sampling space. It turns out that there are theoretical grounds to prefer BCa in general. It is”second-order accurate”, meaning that it converges faster to the correct coverage. Unless you have a reason to do otherwise, make sure to perform a sufficient number of bootstrap replicates (a few thousand is usually not too computationally intensive) and go with reporting BCa intervals.”
- Packages
- {rsample}
- {DescTools::BootCI}
bootandboot.ci- {ebtools::get_boot_ci}
- Steps
- Resample with replacement
- Calculate statistic of resample
- Store statistic
- Repeat 10K or so times
- Calculate mean, sd, and quantiles for CIs across all collected statistics
- Bayesian Bootstrapping (aka Fractional Weighted Bootstrap)
Misc
- Notes from
- Packages
- {fwb}
- Also has two other distributions from which to draw the weights. These are “beta” for a Beta(1/2, 3/2) distribution and “power” for a Beta(√2-1, 1) distribution. These were demonstrated to perform well in small samples by Owen (2025)
- {fwb}
Description
- Doesn’t resample the dataset, but samples a set of weights from the Uniform Dirichlet distribution and computes weighted averages (or whatever statistic)
- Weights sum to ‘n’ but may be non-integers
- Each row gets a frequency weight based on the number of times they appear
- In this way, every row is included in the analysis but given a fractional weight that represents its contribution to the statistic
- In a traditional bootstrap, some rows of data may not be sampled and therefore excluded from the calculation of the statistic
- Particularly useful with rare events, where a row excluded from a traditional bootstrap sample might cause the whole estimation to explode (e.g., in a rare-events logistic regression where one sample has no events!)
- In a traditional bootstrap, some rows of data may not be sampled and therefore excluded from the calculation of the statistic
Should be faster and consume less RAM
Python implementation
def classic_boot(df, estimator, seed=1): df_boot = df.sample(n=len(df), replace=True, random_state=seed) estimate = estimator(df_boot) return estimate def bayes_boot(df, estimator, seed=1): np.random.seed(seed) w = np.random.dirichlet(np.ones(len(df)), 1)[0] result = estimator(df, weights=w) return result from joblib import Parallel, delayed def bootstrap(boot_method, df, estimator, K): r = Parallel(n_jobs=8)(delayed(boot_method)(df, estimator, seed=i) for i in range(K)) return r s1 = bootstrap(bayes_boot, dat, np.average, K = 1000)
Descriptive Statistics
Misc
- Packages
- {directadjusting} - Compute estimates and confidence intervals of weighted averages quickly and easily
- {MADtests} - Hypothesis Tests and Confidence Intervals for Median Absolute Deviations
- {rquest} - Functions to conduct hypothesis tests and derive confidence intervals for quantiles, linear combinations of quantiles, ratios of dependent linear combinations and differences and ratios of all of the above for comparisons between independent samples. Additionally, quantile-based measures of inequality are also considered.
- {wstats} - Weighted versions of common descriptive statistics (variance, standard deviation, covariance, correlation, quantiles).
Means and Medians
Geometric Mean
summarize_revenue <- function(tbl) { tbl %>% summarize(geom_mean_revenue = exp(mean(revenue)), n = n()) }
Proportions
- Sample Size
- {bssbinom} - Bayesian Sample Size for Binomial Proportions
- Variance of a proportion
Assume that p applies equally to all n subjects
\[\sigma^2_p = \frac{p(1-p)}{n}\]
- Example
- Sample of 100 subjects where there are 40 females and 60 males
- 10 of the females and 30 of the males have the disease
- Marginal estimate of the probability of disease is (30+10)/100 = 0.4
- Variance of the estimator assuming constant risk (i.e. assuming risk for females = risk for males)
- (prob_of_disease × prob_not_disease) / n = (0.4 × 0.6) / 100 = 0.0024
- p = (10 + 30) / 100 = 0.40
- (prob_of_disease × prob_not_disease) / n = (0.4 × 0.6) / 100 = 0.0024
- Example
Assume p depends on a variable (e.g. sex)
\[\sigma^2_p = \frac{p^2_{1,n} \cdot p_1(1-p_1)}{n_1} + \frac{p^2_{2,n} \cdot p_2(1-p_2)}{n_2}\]
- Example
- Description same as above
- Adjusted marginal estimate of the probability of disease is
- (prop_female × prop_disease_female) + (prop_male × prop_disease_male)
- (0.4 × 0.25) + (0.6 × 0.5) = 0.4
- Same marginal estimate as before
- Variance of the estimator assuming varying risk (i.e. assumes risk for females \(\neq\) risk for males)
- 1st half of equation:
- prop_female2 × (prop_disease_female × prop_not_disease_female) / n_female = [0.42 × (0.25 × 0.74) / 40]
- 2nd half of equation
- prop_male2 × (prop_disease_male × prop_not_disease_male) / n_male = [0.62 × (0.5 × 0.5) / 60]
- 1st half + 2nd half = 0.00224
- Variance is smaller than before
- 1st half of equation:
- Example
- CIs
Packages:
- {binomCI} - 12 confidence intervals for one binomial proportion or a vector of binomial proportions are computed
Jeffrey’s Interval
# probability of event # n_rain in the number of events (rainy days) # n is the number of trials (total days) mutate(pct_rain = n_rain / n, # jeffreys interval # bayesian CI for binomial proportions low = qbeta(.025, n_rain + .5, n - n_rain + .5), high = qbeta(.975, n_rain + .5, n - n_rain + .5))
Skewness
- Packages:
- {moments} - Standard algorithm
- {e1071} - 3 alg options
- {DescTools::Skew} - Same algs but with bootstraps CIs
- From the paper referenced in e1071, b1 (type 3) is better for non-normal population distributions and G1 (type 2) is better for normal population distributions
- Symmetric: Values between -0.5 to 0.5
- Moderated Skewed data: Values between -1 and -0.5 or between 0.5 and 1
- Highly Skewed data: Values less than -1 or greater than 1
- Relationship between Mean and Median under different skewness

Kurtosis
A high kurtosis distribution has a sharper peak and longer fatter tails, while a low kurtosis distribution has a more rounded peak and shorter thinner tails.
Types

- Mesokurtic: kurtosis = ~3
- Examples: normal distribution. Also binomial distribution when p = 1/2 +/- sqrt(1/12)
- Leptokurtic: This distribution has fatter tails and a sharper peak. Excess kurtosis > 3
- Examples: Student’s t-distribution, Rayleigh distribution, Laplace distribution, exponential distribution, Poisson distribution and the logistic distribution
- Platykurtic: The distribution has a lower and wider peak and thinner tails. Excess kurtosis < 3
- Examples: continuous and discrete uniform distributions, raised cosine distribution, and especially the Bernoulli distribution
- Excess Kurtosis is the kurtosis value - 3
- Mesokurtic: kurtosis = ~3
Understanding CI, SD, and SEM Bars
- article
- P-values test whether the sample means are different from each other
- sd bars: Show the population spread around each sample mean. Useful as predictors of the range of new sample.
- Never seen these and it seems odd to mix a sample statistic with a population parameter and that the range is centered on the sample mean (unless the sample size is large I guess).
- s.e.m. is the “standard error of the mean” (See Terms)
- In large samples, the s.e.m. bar can be interpreted as a 67% CI.
- 95% CI ≈ 2 × s.e.m. (n > 15)
- Figure 1
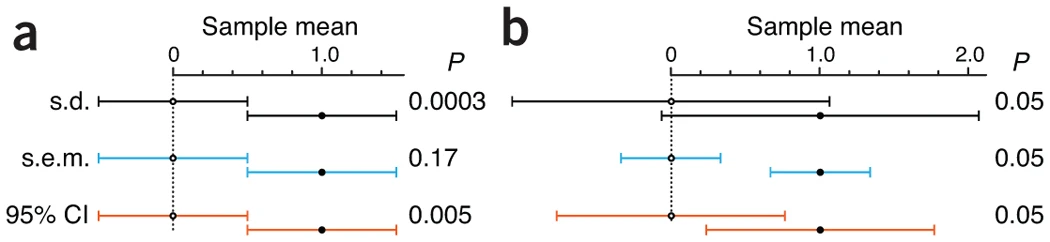
- Each plot shows 2 points representing 2 sample means
- Plot a: bars of both samples touch and are the same length
- sem bars intepretation: Commonly held view that “if the s.e.m. bars do not overlap, the difference between the values is statistically significant” is NOT correct. Bars touch here but don’t overlap and the difference in sample means is NOT significant.
- Plot b: p-value = 0.05 is fixed
- sd bar interpretation: Although the means differ, and this can be detected with a sufficiently large sample size, there is considerable overlap in the data from the two populations.
- sem bar intepretation: For there to be a significant difference in sample means, sem bars have to much further away from each other than there just being a recognizable space between the bars.
- Figure 2
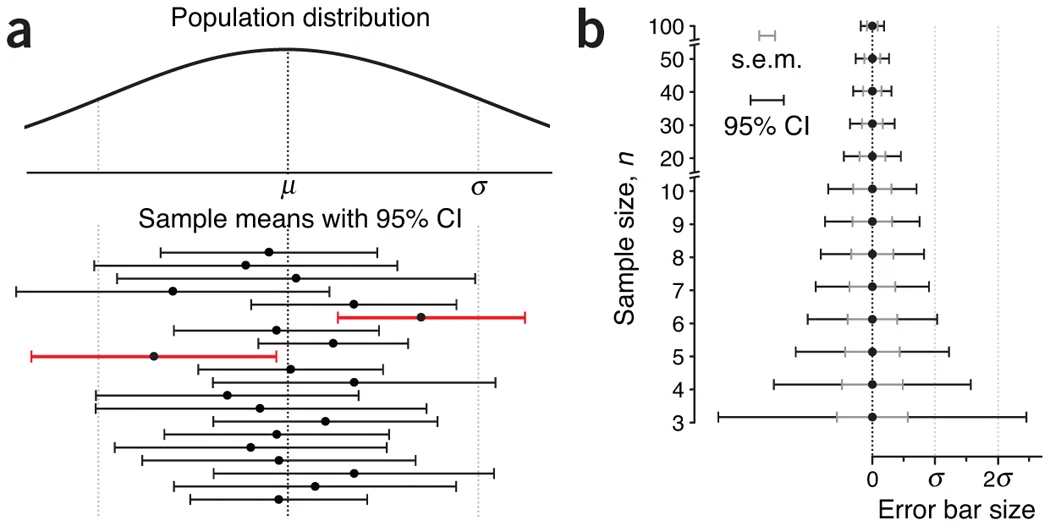
- Plot a: shows how a
- 95% CI captures the population mean 95% of the time but as seen here, only 18 out of 20 sample CIs (90%) contained the population mean (i.e. this is an asymptotic claim)
- A common misconception about CIs is an expectation that a CI captures the mean of a second sample drawn from the same population with a CI% chance. Because CI position and size vary with each sample, this chance is actually lower.
- Plot b:
- Hard to see at first but the outer black bars are the 95% CI and the inner gray bars are the sem.
- Both the CI and sem shrink as n increases and the sem is always encompassed by the CI
- Plot a: shows how a
- Figure 3
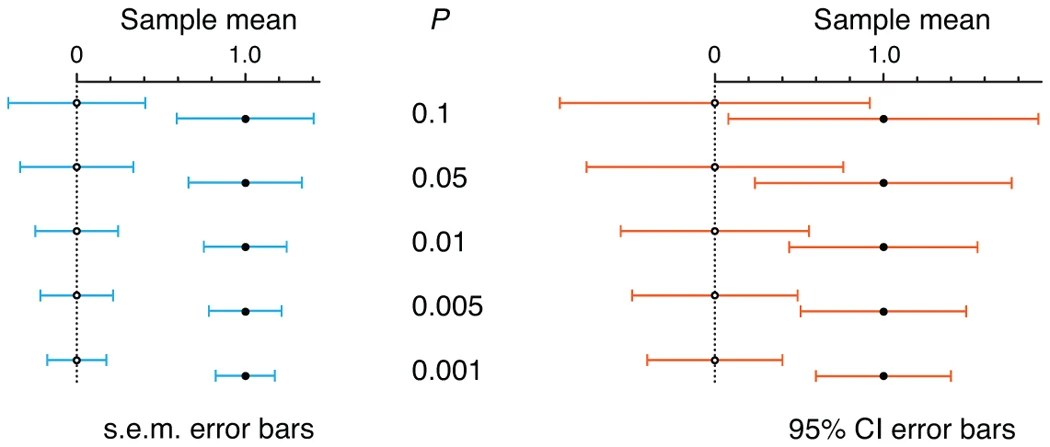
- sem bars must be separated by about 1 sem (which is half a bar) for a significant difference to be reached at p-value = 0.05
- 95% CI bars can overlap by as much as 50% and still indicate a significant difference at p-value = 0.05
- If 95% CI bars just touch, the result is highly significant (P = 0.005)
P-Values
Misc
- Packages
- {boot.pval} - Computation of bootstrap p-values through inversion of confidence intervals, including convenience functions for regression models.
- Linear models fitted using
lm, - Generalised linear models fitted using
glmorglm.nb, - Nonlinear models fitted using
nls, - Robust linear models fitted using
MASS::rlm, - Ordered logistic or probit regression models fitted (without weights) using
MASS:polr, - Linear mixed models fitted using
lme4::lmerorlmerTest::lmer, - Generalised linear mixed models fitted using
lme4::glmer. - Cox PH regression models fitted using
survival::coxph(usingcensboot_summary). - Accelerated failure time models fitted using
survival::survregorrms::psm(usingcensboot_summary). - Any regression model such that:
residuals(object, type="pearson")returns Pearson residuals;fitted(object)returns fitted values;hatvalues(object)returns the leverages, or perhaps the value 1 which will effectively ignore setting the hatvalues. In addition, thedataargument should contain no missing values among the columns actually used in fitting the model.
- Linear models fitted using
- {pplot} - Chronological and Ordered p-Plots for Empirical Data
- The p-plot visualizes the evolution of the p-value of a significance test across the sampled data. It allows for assessing the consistency of the observed effects, for detecting the presence of potential moderator variables, and for estimating the influence of outlier values on the observed results
- {boot.pval} - Computation of bootstrap p-values through inversion of confidence intervals, including convenience functions for regression models.
P-Value Function
- Notes from https://ebrary.net/72024/health/value_confidence_interval_functions
- {concurve} creates these curves
- Gives a more complete picture than just stating the p-value (strength and precision of the estimate)
- Shows level of precision of the point estimate via shape of the curve
- narrow-based, spikey curves = more precise
- Visualizes strength of the effect along the x-axis. Helps in showing “significant” effect is not necessarily a meaningful effect.
- Shows level of precision of the point estimate via shape of the curve
- Shows other estimate(s) that are also consistent with that p-value
- Shows p-values associated with other estimates for the Null Hypothesis
- see the end of the article for discussion on using this fact in an interpretation context
- The P-value function is closely related to the set of all confidence intervals for a given estimate. (see example 3)
- Example 1
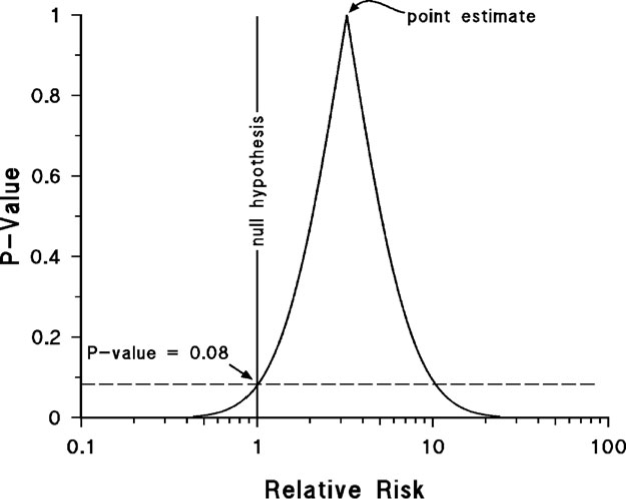
- p-value of the point estimate is (always?) 1 which says, “given a null hypothesis = <pt est> is true (i.e. the true risk ratio = <pt est>), the probability of seeing data produce this estimate or this estimate with more strength (ie smaller std error) is 100%.”
- I.e. the pt est is the estimate most compatible with the data.
- This pval language is mine. The whole “this data or data more extreme” has never sit right with me. I think this is more correct if my understanding is right.
- The pval for the data in this example is at 0.08 for a H0 of 1. So unlikely, but typically not unlikely enough in order to reject the null hypothesis.
- A pval of 0.08 is identical for a pt est = 1 or pt est = 10.5
- Wide base of the curve indicates the estimate is imprecise. There’s potentially a large effect or little or no effect.
- p-value of the point estimate is (always?) 1 which says, “given a null hypothesis = <pt est> is true (i.e. the true risk ratio = <pt est>), the probability of seeing data produce this estimate or this estimate with more strength (ie smaller std error) is 100%.”
- Example 2
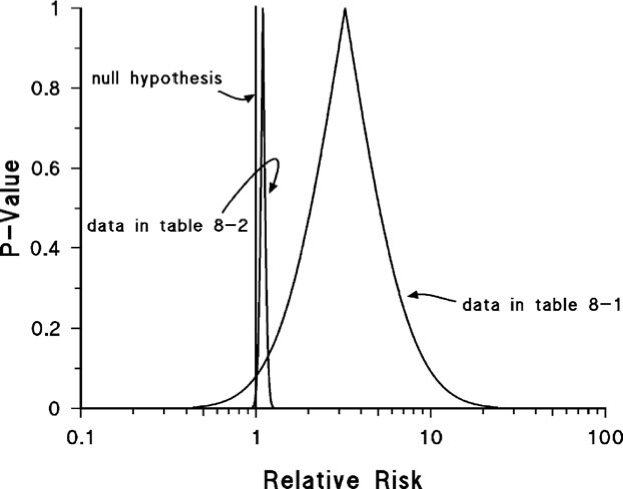
- more data used for the second curve which indicates a precise point estimate.
- pt est very close to H0
- pval = 0.04 (not shown in plot)
- So the arbitrary pval = 0.05 threshold is passed and says a small effect is probably present
- Is that small of an effect meaningful even if it’s been deemed statistically present?
- In this case a plot with the second curve helps show that “statistically significant” doesn’t necessarily translate to meaningful effect
- Example 3
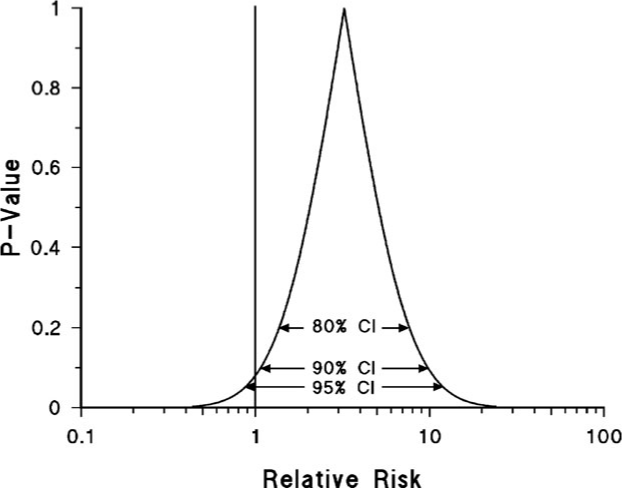
- The different confidence intervals reflect the same degree of precision (i.e. the curve width doesn’t change when moving from one CI to another).
- The three confidence intervals are described as nested confidence intervals. The P-value function is a graph of all possible nested confidence intervals for a given estimate, reflecting all possible levels of confidence between 0% and 100%.
Multivariate
Misc
- Also see Regression, Multivariate
- Multivariate Analysis (MVA) is used to address situations where multiple measurements are made on each experimental unit and the relations among these measurements and their structures are important
- It’s concerned with the joint analysis of multiple dependent variables
- If the data were all independent columns, then the data would have no multivariate structure and you could just do univariate statistics on each variable (column) in turn. Multivariate statistics means we are interested in how the columns covary.
- Packages
- CRAN Task View: Compositional Data Analysis
- {Compositional} - Compositional Data Analysis
- Regression, classification, contour plots, hypothesis testing and fitting of distributions for compositional data are some of the functions included. We further include functions for percentages (or proportions).
- Notes from Energy Based Equality of Distributions Testing for Compositional Data
aeqdist.etest- Performs the \(\alpha\)–EBT, energy-based test for equality of distributions of Euclidean compositional data. The \(\alpha\) transformation adds power to the test as compared to RBPTdptest- Performs the RBPT, random projections-based test for equality of distributions of Euclidean compositional data.- Both tests achieve to the correct Type I error rate, but the \(\alpha\) transformation adds power to the \(\alpha\)–EBT test as compared to RBPT. The downside is \(\alpha\)–EBT’s computational complexity, memory requirements, especially with large data sets.
- {PlotNormTest} - Graphical Univariate/Multivariate Assessments for Normality Assumption
- {coda.plot} - Provides a collection of easy-to-use functions for creating visualizations of compositional data using ‘ggplot2’
- {rrcovNA} - Robust Location and Scatter Estimation and Robust Multivariate Analysis with High Breakdown Point for Incomplete Data (missing values)
- Resources
Terms
- Compositional Data - Positive multivariate data where the sum of each vector is the same constant, usually 1
- Surprisingly the Dirchlet distribution is not, statistically, rich and flexible enough to capture many types of variabilities (especially curvature) of compositional data
- Usages (source)
- Sedimentology - Samples were taken from an Arctic lake and their composition of water, clay and sand were the quantities of interest.
- Oceanography - Studies involving Foraminiferal (a marine plankton species) compositions at 30 different sea depths
- Hydrochemistry - Regression analysis is used to draw conclusions about anthropogenic and geological pollution sources of rivers in Spain.
- Econmomics - The percentage of the household expenditure allocated to different products
- Political Science - A regression analysis used to explain or predict how geographic distributions of electoral results depend upon economic conditions, neighborhood ethnic compositions, campaign spending, and other features of the election campaign or aggregate areas.
- Depth - A measure of centrality. A function assigns to each point \(X \in \mathcal{X}\) (where \(\mathcal{X}\) is a metric space) a measure of centrality \(D(X; P_x)\) with respect to a given probability distribution \(P_X\) on \(\mathcal{X}\) . (source)
- The larger \(D(X; P_X)\) is, the more centrally located \(X\) is with respect to the probability mass of \(P_X\) (empirical distribution is used in practice which is denoted, \(P_n\)). And, vice versa, points with small depths can be seen as outlying in the view of \(P_X\).
- Metric Depth means analyzing data the belong in a arbitrary metric space, \(\mathcal{X}\), since many modern applications produce data that are inherently non-Euclidean (functions, compositions, trees, graphs, rotations, positive-definite matrices, etc.), known also as object data. (source)
- Properties
- Depend on the data only through the metric \(d\), making them applicable to any form of object data, regardless in which specific metric space they live
- If \((\mathcal{X},d)\) is chosen to be a Euclidean space, then the original Euclidean version of the depth is recovered, showing that these metric depth functions are indeed “true” generalization of their classical counterparts.
- Properties
- Deepest Point is the object which has the maximal depth value \(D(X;P_n)\) and serves as the analogue of sample mean for object data.
Depth
- Misc
- Usage
- A robust multivariate sample mean can be calculated by finding the Deepest Point and therefore difference-in-means can be calculated and tested.
- Outliers can be detected by determining the “shallow”(?) points (i.e. minimum depth values)
- A distance metric must be chosen for metric depth measures. For example, euclidean distance (\(\mathcal{L}_2\)) for euclidean data or arc-length for spherical data (e.g. geospatial)
- Most Depth measures are robust to outliers but not location measures like the Fréchet mean which is a generalization of the concept of average and a classical estimator of location for object data (source)
- Usage
- Oja Median
- Multivariate median for Euclidean data
- Computationally intensive
- {OjaNP} - Package has been archived by CRAN but can be downloaded through the link to the journal article/vignette.
- Metric Oja Depth
- Paper, Code
- Also includes code for metric half-space depth, metric lens depth, and metric spatial depth
- Paper performs a comparison between 2 forms of Oja Depth, along with metric half-space depth, metric lens depth, and metric spatial depth. In the Discussion, it provides guidelines for choosing which one.
- Computationally intensive
- Paper Example
- 35 Canadian weather stations are divided into two groups — “the eastern and western coastal areas, and the northern and central southern areas — and our research question is: Is there a significant difference in the typical temperature curves between these two groups?”
- Paper, Code
- Metric Spatial Depth
- Paper, Code - Reading the function code by itself doesn’t make it clear to me on how to use it. Maybe by reading the paper, it would be clear. The Oja Depth paper has the code for the analysis and simulation used in that paper, and this metric is also used. It might be faster to read that paper and it’s code.
- Paper Example: Outlier Detection
- Wasserstein Spatial Depth
Miscellaneous Tests
- Hettmansperger-Norton Test for Patterned Alternatives in k-Sample Problems (Paper)
Tests for various trends across groups
Example: Suppose that we suspect that with increasing age, one’s ability to perform a certain job tends to improve, but that after some point advancing age tends to mean diminishing performance. Suppose that N employees are grouped by age into k groups and employees’ job performances are ranked from 1 to N. Viewing performance as the dependent variable, the particular pattern of increasing group locations followed by decreasing group locations is called an umbrella (pattern).
Example: {pseudorank::hettmansperger_norton_test}
# create some data, please note that the group factor needs to be ordered df <- data.frame( data = c(rnorm(40, 3, 1), rnorm(40, 2, 1), rnorm(20, 1, 1)), group = c(rep(1,40), rep(2,40), rep(3,20)) ) df$group <- factor(df$group, ordered = TRUE) # you can either test for a decreasing, increasing or custom trend hettmansperger_norton_test(df$data, df$group, alternative="decreasing") hettmansperger_norton_test(df$data, df$group, alternative="increasing") hettmansperger_norton_test(df$data, df$group, alternative="custom", trend = c(1, 3, 2))- trend = c(1,3,2) describes an umbrella pattern (like the first example). 1 increases to 3. Then, 3 decreases to 2
- If there were five groups, the umbrella trend would be specified by trend = c(1, 2, 5, 4, 3).